SRST2-ARGannot抗药性基因数据库之深度解析
在细菌基因组流行病学(genomic epidemiology)领域,SRST2是一个通过细菌全基因组测序读片段(reads)和参考基因数据库来快速检测目标基因的软件1。SRST2-ARGannot是一个由Kathryn Holt实验室创建并维护、符合SRST2输入格式的ARG-ANNOT数据库。目前最新的版本是r3(revision 3,ARGannot_r3.fasta)。我作为SRST2-ARGannot r2版本的整理者[r3版本仅在r2基础上增加了抗粘菌素(colistin)的数个msr基因],在长期使用SRST2的过程中积累了丰富的经验。在此,我对SRST2-ARGannot数据库作一个深入解析,希望能够对从事相关分析的读者有所帮助。
1. 细菌遗传学基本概念:基因座、基因与等位基因
厘清基因座(locus)、基因(gene)与等位基因(allele)这三个遗传学中的基本概念,是正确理解SRST2-ARGannot数据库和SRST2工作原理的基础。与人类基因组相比,细菌与古生菌(Archaea)的基因组由于普遍存在大量的可移动原件(mobile genetic elements)和可移动基因(mobile genes),在结构上存在很大的动态性。这三个概念的外延和相互关系也因此变得复杂。在本文中,我仅讨论这几个概念在细菌基因组中的内涵与外延。同时,请读者注意,文中的结论可以延伸到古生菌的基因组。
1.1. 基因座
又称“位点”,指DNA分子上特定的性状决定因子的物理位置。在细菌基因组中,基因座不仅可以存在于染色体上,还可以存在于可移动元件(比如质粒)上;可以由多个基因组成,并且不一定为所有同物种的个体共享[例如获得性荚膜合成位点(capsule synthesis loci)5, 6——更确切地说,应该是荚膜合成基因(组)岛(genomic island)]。与人类基因组不同,因为细菌基因组结构的动态性,相同基因座的具体位置在同一细菌物种的不同基因组间可能存在差异,所以“物理位置”这一概念内涵被弱化。
A locus is a spot or “address” on a chromosome at which a gene for a particular trait is located in all members of a species. It can also refer to the location of a mutation or other genetic marker.3, 4
A locus is the specific physical location of a gene or other DNA sequence on a chromosome, like a genetic street address. — (US) National Human Genome Research Institute (NHGRI)
1.2. 基因
人们对基因的定义经历了一百余年的发展过程,并且仍然在争议中演变。总结起来,人们当前对基因这一概念的内涵达成的共识包括:(1)单个基因是位于DNA上某特定基因座的核酸序列(即拥有确定且唯一的位置,否则视为两个相同或不同的基因);(2)编码蛋白质或非编码性RNA(ncRNA),或直接调节转录过程;(3)产物的序列可以不唯一,功能可以不同,但序列须重叠。对缺少内含子(intron)的细菌基因来说,产物常常只有一个。在2007年,Gerstein等人基于基因的功能性产物(functional products),对基因给出了如下定义:
A gene is a union of genomic sequences encoding a coherent set of potentially overlapping functional products7.
这一概念的延伸见下图(引自Gerstein等人论文7的图5):

图1:应用Gerstein等人提出的基因定义的示范
按照该定义,非同源、不相重叠却又同为细菌抗链霉素基因的strA与strB是两个不同的基因(其基因符号符合Demerec等人提出的方案9)。此外,这个定义还暗示,以前按照开放阅读框ORF(open reading frame)定义的重叠基因应该被视作一个融合基因(fusion gene or hybrid gene)。该融合基因可能产生两种部分序列重叠的功能性产物(例外的情况如沙门氏菌SGI1基因岛中的融合基因qacEΔ1-sul1。其原有的qacE1基因残缺,不再形成完整的功能性产物)。值得一提的是,Gerstein等人还补充道,基因的内涵还必须包括,所有重叠的产物来自同一基因座(即物理同源)。因此,尽管旁系同源的两个基因座的产物可能高度重叠,但这两个基因座中的DNA序列应当被视作两个基因。
在2017年,Portin和Wilkins对“基因”这一概念给出了如黑格尔哲学一般复杂的定义:
A gene is a DNA sequence (whose component segments do not necessarily need to be physically contiguous) that specifies one or more sequence-related RNAs/proteins that are both evoked by genetic regulatory networks (GRNs) and participate as elements in GRNs, often with indirect effects, or as outputs of GRNs, the latter yielding more direct phenotypic effects8.
然而,按照Gerstein等人在其论文7中的主张,基因的定义应当方便实践,Portin和Wilkins对基因这一概念做出的限定显得过于抽象,从而缺乏可操作性。在研究细菌基因组的文献中,我们还经常能见到更早的,并且依然具有实践意义的基因定义。具有代表性的有:
The terms “locus” and “gene” will be used interchangeably to refer to a specific sequence of nucleotides governing the sequence of amino acids in a specific polypeptide (or the sequence of nucleotides in a specific RNA molecule). Nucleotide sequences which themselves may not be transcribed, but which govern the punctuation or regulation of transcription, are also referred to as “loci”9. — Demerec et al. (1966)
显然,在当时的作者看来,基因和基因座是同义词。不过,在我的理解中,基因座强调的是基因的物理位置,而基因强调的是位于那个位置的核酸序列。由于在细菌基因组中,获得性基因的位置常常不固定,而核酸序列才是产物功能的决定因素,所以我倾向于将Demerec等人对基因的定义限定于序列层次而不是基因座层次。不过,Demerec等人的定义符合上世纪60年代,人们把基因视作编码某特定功能性产物的一段DNA序列的普遍观点7。
1.3. 等位基因
不考虑单核苷酸多态性(SNP)的情况,同一基因的同源核酸序列互为等位基因。该定义来自allele的词源并具有如下三点内涵:(1)共同的基因座(非旁系同源);(2)序列同源,也就是按照经典遗传学的表述,可以发生染色质互换(crossover);(3)等位基因间的对等性(可互换)。
The different forms of a locus brought about by such mutations are called alleles9. — Demerec et al. (1966)
在实践中,显著的序列相似性是分化(divergence)程度较低的序列同源的必要条件。在BLAST的结果中,该显著性由序列相似度(Score计分)的E-value反映 [与被查询序列(query sequence)的长度和数据库大小有关]。此外,非旁系同源与序列同源这两个约束条件说明,等位基因属于同一直系同源(序列)群(orthologous group)。
1.4. 其他观点
在以上讨论的内容之外,我很欣赏BIGSdb数据库平台的作者Keith Jolley给出的操作性定义:
Loci are regions of the genome that are identified by similarity to a known sequence. They can be defined by DNA or peptide sequence. They are often complete coding sequences (genes), but may represent gene fragments (such as used in MLST), antigenic peptide loops, or indeed any sequence feature.
Alleles are instances of loci. Every unique sequence, either DNA or peptide depending on the locus, is defined as a new allele and these are defined in a sequence definition database, where they are given an allele identifier.
2. 理解SRST2-ARGannot数据库
本数据库的格式和构建方法可见SRST2的官方手册。在此,我对手册中指出的序列名称格式进行展开:
>[clusterUniqueIdentifier]__[clusterSymbol]__[alleleSymbol]__[alleleUniqueIdentifier]
正如我在本文第1节中所指出,细菌基因组的动态性为基因和基因座的定义带来了复杂性和特殊性。同时,因为人们对序列同源性的判断并没有一个绝对标准,所以往往难以在一组相似序列是否属于同一基因的等位基因这个问题上达成共识[对趋同演化(convergence evolution / homoplasy)可能性的测试是其中的又一个难点]。这样的困境,使人们常常混用细菌基因和等位基因这两个概念。比如,在文献中被逐一称为基因的细菌抗药性决定因子_bla_TEM-x(x为数字)系列实则同属等位基因。
2.1. 序列聚类
鉴于细菌基因命名系统(nomenclature)的混乱现状,SRST2的设计者采用了一种通用的序列组织方式以回避判断参考序列是否为等位基因的繁琐和不确定性:将数据库中的序列聚类,然后在SRST2对每个样本基因组呈现最终的分析结果时,仅报告每个类别(序列簇,sequence cluster)下,最接近样本基因的参考序列(在数据库和结果报告中被称为“等位基因”)。具体来说,我们有两种聚类方式:
- 按已知的基因和等位基因聚类(理想情况);
- 当关于基因和等位基因的信息不完整时,按照序列相似性聚类。
由于细菌抗药性基因的命名属于第二种情况,SRST2-ARGannot数据库基于80%核酸序列相似性(使用CD-HIT-EST计算)定义序列簇。每个簇的符号(clusterSymbol)由代表序列名 + 抗药性类别(缩写为AGly,Bla,等等)组成(并非固定格式)。比如,>205__TEM-1D_Bla__TEM-1D__887表示在序列簇TEM-1D_Bla中的序列TEM-1D__887。在同一簇下,还有序列如TEM-2__889等。特别地,序列编号(如887和889)被附加到传统的等位基因名称上以解决不同序列重名的情况。SRST2-ARGannot数据库的这种组织结构,是原有的ARG-ANNOT数据库(仅按抗药性类别聚类)不具备的。同时,前者排除了后者中的冗余序列(100%相似)。
2.2. 为何要聚类?
ARG-ANNOT和ResFinder数据库并没有对参考序列按照相似性聚类。此外,这两个数据库的官方网站为搜索基因组组装体(genome assembly)提供了BLAST接口。在每次分析结束后,对组装体上每一个抗药性基因座,我们会得到在全数据库范围(而不是单个序列簇范围)内,满足预设阈值的最佳匹配 [ResFinder的最佳匹配判定方法同基于长度计分(Length Score)和相似度百分比(percentage of identity)的MLST分析13, 14]。相比之下,SRST2基于聚类数据库的分析可以被视作在报告单一命中序列的功能之外,提供序列簇层次上的一种归纳。那么,这样的聚类操作是不是必须的呢?
我认为,序列聚类对于在组装体中寻找目标基因的方法(比如BLAST和ResFinder),不是必须的;但对于通过测序的读片段来检测目标基因的方法(比如SRST2)而言,则是必须的。
为什么呢?一方面,如我在本节第一段里提到的,基于组装体的方法可以为每一个抗药性基因座确定最佳的序列比对结果(图2与3)。因为在组装体中,每个基因座的位置是确定的,所以不难获得最匹配的参考序列。
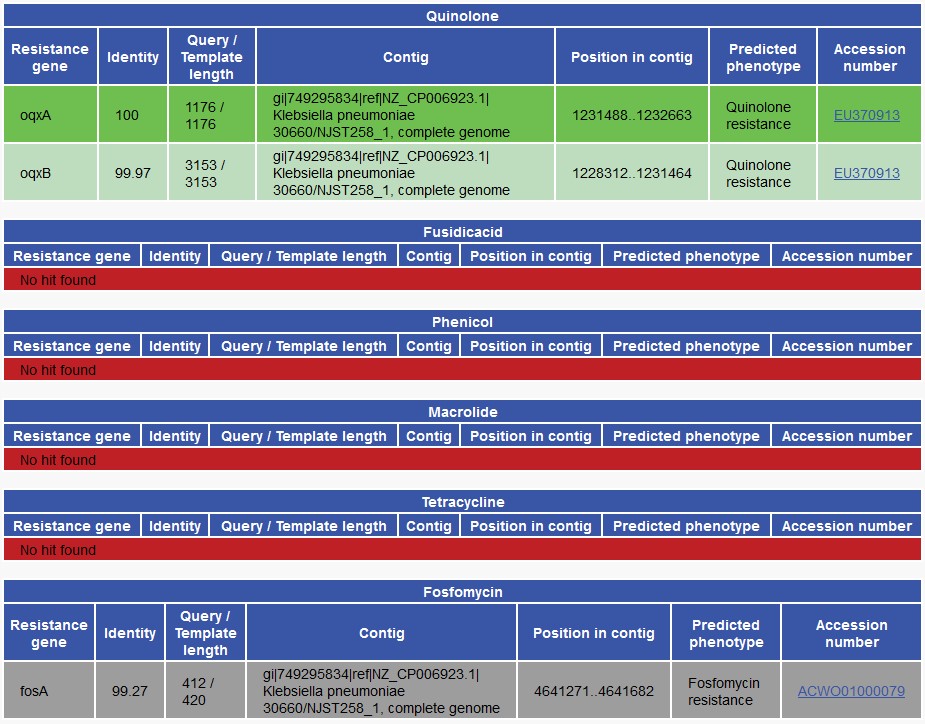
图2:ResFinder输出示例
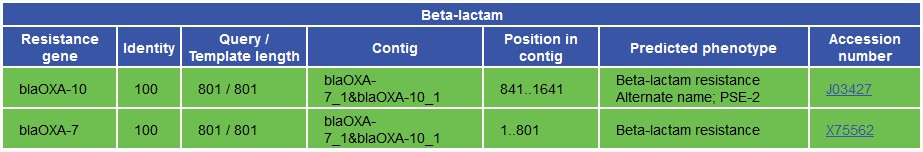
图3:ResFinder对一段包含两个等位基因(blaOXA-7和_bla_OXA-10)的人造序列的分析结果
在另一方面,对于如SRST2这般,将读片段排列到参考序列上的方法,我可以通过一个思想实验来论证序列聚类的必要性。首先,假设在参考数据库中,记录了基因 a 的等位基因a-1,a-2和a-3,以及基因b的等位基因b-1,b-2,b-3和b-4。并且,基因 a 和 b 非同源。同时,假设在某细菌的基因组中,分别有目标基因a和b的等位基因a-1和b-1’,其中b-1’指代最接近b-1的一个等位基因。那么,将该基因组的读片段集合通过SRST2比对到参考数据库后,可以预见序列的相似性会出现如下从高到低的顺序:
- a-1 > a-2, a-3
- b-1 > b-2, b-3, b-4
但是,因为仅通过基因组的读片段,我们并不能知道命中 a-1 和 b-1 参考序列的样本基因是来自于同一个还是两个基因座,所以在不知道 a-1 和 b-1 序列的相似性的情况下,我们无法对这两个命中项进行取舍(舍弃其一,或全部接受)。而如果此时知道 a-1 和 b-1 的相似度,则数据库又回到了经过聚类的形式。也就是说,根据 a-1 和 b-1 的相似性(比如,80%核算相似度与90%序列覆盖度),可能有两种结果:
- 相似:仅报告发现 a-1 ;
- 不相似:报告同时发现 b-1 的一个等位基因和_a-1_ 。
综上所述,对序列进行聚类,是基于读片段来检测目标基因的方法的本质需求。在2.3节的第二点,我们还会看到,对数据库做聚类操作时采用的相似度阈值直接决定基因检测的灵敏度(sensitivity)和特异性(specificity)。
2.3. 存在的问题
SRST2-ARGannot数据库解决了序列冗余和重名的问题,并且提供了序列的聚类信息。但是,当前的数据库存在两个突出的问题:
- 在呈现结果时具有误导性的名称:按照前面的分析,当SRST2的参考数据库按照序列的相似性而非确定的基因来聚类时,每个类别(对应SRST2输出表格中的一列)就不应该被视作基因,无论该类别的符号如何。这样的设计,为数据库的组织和结果解释带来了灵活性(用户可以根据需要定制不同的聚类方式并分配类别名称),还反映了SRST2的工作原理。但是,由于SRST2的术语表和结果格式预设了2.1节中第一点这种理想情况,我们在SRST2的软件文档中会看到序列簇被称作”基因“,而每个簇的成员序列被称为”等位基因“。特别地,在SRST2的”fullgene“报告中,每个簇被明确地视作一个基因,其下还有最匹配的等位基因的信息。然而,当真实的情况是2.1节中第二点那样的非理想状态时,这些命名具有强烈的误导性:研究者可能不会意识到序列间存在相似性。比如,在一篇综述12中,作者将_bla_OXA-1、blaOXA-2、blaOXA-7和_bla_OXA-10视为四个抗药性基因。真实的情况却是,尽管_bla_OXA-1和_bla_OXA-2这两个序列都编码D类内酰胺酶(beta-lactamases),但因为这两段序列不具有任何同源性(根据BLASTn的结果),它们属于不同基因。相反,抗药性决定因子_bla_OXA-7与_bla_OXA-10因为具有相同的序列长度(801 bp)和96%的核苷酸相似性(E-value = 0),而应该被视作同一基因的不同等位基因。类似的情形还见于_bla_OXA-48和_bla_OXA-181这一对等位基因(798 bp,94%核苷酸相似度)——作者同样把它们视为两个不同的基因。
- 武断的聚类依据:一方面,为什么按照80%(或其它给定阈值)核酸相似性定义的一个序列簇就可以总是可以(或者不可以)被当作一个基因呢?我们事实上并没有对SRST2-ARGannot数据库的序列相似性做过宏观的调查(例如,可以借助于网络图来表示序列间的相似性)。另一方面,从2.2节的结论得到的一个推论是,如果我们盲目提高聚类时采用的相似度阈值,比如提高到95%甚至100%,我们可能引入假阳性结果(例如,将阈值提高到100%,则数据库回归到ARG-ANNOT和ResFinder的状态——每个序列就是一个类——从而无法通过读片段获得准确的基因检测结果,因为同一个样本基因会匹配到所有跟它相似且彼此相似的类上);反之,如果盲目降低聚类时的相似度阈值,比如下调至50%或更低,我们将引入假阴性结果(有一定相似度的序列簇被合并为一个)。因此,我们应该理性选择这个阈值。
- 序列簇的符号不一定对应被广泛接受的基因符号:在分析SRST2的结果时,我们需要人工地检查簇的符号是否对应被广泛认可的基因符号。Moradigaravand、Martin、Peacock 和Parkhill发表于2017年的一篇文章中的图5示范了人工调整后的基因符号11。
参考文献
- Inouye, M., Dashnow, H., Raven, L.-A., Schultz, M., Pope, B., Tomita, T., … Holt, K. (2014). SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Medicine, 6(11), 90. Retrieved from http://genomemedicine.com/content/6/11/90.
- Gupta, S. K., Padmanabhan, B. R., Diene, S. M., Lopez-Rojas, R., Kempf, M., Landraud, L., & Rolain, J. M. (2014). ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother, 58(1), 212–220. https://doi.org/10.1128/aac.01310-13.
- Turner J.R. (2013) Locus (Genetics). In: Gellman M.D., Turner J.R. (eds) Encyclopedia of Behavioral Medicine. Springer, New York, NY.
- Brown, S. M. (2009). Essentials of medical genomics (2nd ed.). Hoboken, NJ: Wiley-Blackwell.
- Bartley, S. N., Mowlaboccus, S., Mullally, C. A., Stubbs, K. A., Vrielink, A., Maiden, M. C., … Kahler, C. M. (2017). Acquisition of the capsule locus by horizontal gene transfer in Neisseria meningitidis is often accompanied by the loss of UDP-GalNAc synthesis. Scientific reports, 7, 44442. doi:10.1038/srep44442.
- Wyres, K. L., Wick, R. R., Gorrie, C., Jenney, A., Follador, R., Thomson, N. R., & Holt, K. E. (2016). Identification of Klebsiella capsule synthesis loci from whole genome data. Microbial genomics, 2(12), e000102. doi:10.1099/mgen.0.000102.
- Gerstein, M. B., Bruce, C., Rozowsky, J. S., Zheng, D., Du, J., Korbel, J. O., … Snyder, M. (2007). What is a gene, post-ENCODE? History and updated definition. Genome Research , 17(6), 669–681. https://doi.org/10.1101/gr.6339607.
- Portin, P., & Wilkins, A. (2017). The Evolving Definition of the Term “Gene.” Genetics, 205(4), 1353 LP – 1364. https://doi.org/10.1534/genetics.116.196956.
- Demerec, M., Adelberg, E. A., Clark, A. J., & Hartman, P. E. (1966). A Proposal for a Uniform Nomenclature in Bacterial Genetics . *Genetics, 54(*1), 61–76. Retrieved from http://www.ncbi.nlm.nih.gov/pmc/articles/PMC1211113/.
- Sequence homology, Wikipedia, https://en.wikipedia.org/wiki/Sequence_homology Retrieved on 30 Nov 2019.
- Moradigaravand, D., Martin, V., Peacock, S. J., & Parkhill, J. (2017). Population structure of multidrug resistant Klebsiella oxytoca within hospitals across the UK and Ireland identifies sharing of virulence and resistance genes with K. pneumoniae. Genome Biology and Evolution, 9(3), 574–584. https://doi.org/10.1093/gbe/evx019.
- Wyres, K. L., & Holt, K. E. (2016). Klebsiella pneumoniae Population Genomics and Antimicrobial-Resistant Clones. Trends in Microbiology, 24(12), 944–956. https://doi.org/http://dx.doi.org/10.1016/j.tim.2016.09.007.
- Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., … Larsen, M. V. (2012). Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother, 67(11), 2640–2644. https://doi.org/10.1093/jac/dks261.
- Larsen, M. V, Cosentino, S., Rasmussen, S., Friis, C., Hasman, H., Marvig, R. L., … Lund, O. (2012). Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. Journal of Clinical Microbiology, 50(4), 1355–1361. https://doi.org/10.1128/JCM.06094-11.